|
AUTOSOMAL DOMINANT
POLYCYSTIC DISEASE OF THE KIDNEY
(ADPCDK) |
· ADPCDK is an autosomal dominant condition, with almost 100% penetrance, that usually presents in the fourth decade of life but may present in the fetus (1,2).
· Adult polycystic kidney disease or DPKD is the most common cystic kidney disease and is responsible for 10% to 12% of patients on chronic dialysis.
· ADPKD is rarely seen in utero because less than 5% of the nephrons are cystic antenatally. There are normal collecting tubules and nephrons interspersed among abnormal areas.
· Abnormalities of chromosomes 16p and 4q have been reported.
·
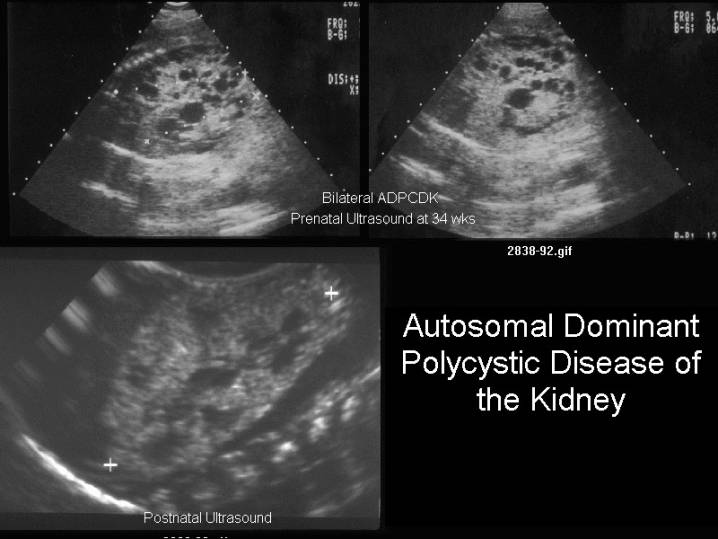
In reported fetal cases, the kidneys were
enlarged and echogenic with cysts seen in the cortex
and medulla. Renal cysts are rarely seen in normal children; hence, when
imaged, although at times part of some syndromes, one must consider the
possibility of ADPKD. Analysis of the
ULTRASOUND |
- Usually both kidneys are enlarged and hyperechoic.
- Multiple renal cysts (unilateral or bilateral).
- Cysts are small and of uniform shape, representing dilated nephrons and not tubules (as in the autosomal recessive form).
- Amniotic fluid volume is normal.
- Hepatic cystic disease (non-obstructive biliary ectasia) occurs occasionally (not usually a feature in the fetus).
- Pancreatic cystic disease (usually not a feature in the fetus).
|
|
REFERENCES |
- Main D,
- Zerres K, Weiss H, Bulla M. Prenatal diagnosis of an early manifestation of autosomal dominant-type polycystic kidney disease. Lancet 1982;2:988.