|
AUTOSOMAL RECESSIVE
POLYCYSTIC DISEASE OF THE KIDNEY
(ARPCDK) |
ARPCDK is a rare condition (1:50,000 infants) that usually presents
in the fetus around 20 weeks gestation (1) but may occasionally not present
until after delivery (1) (neonatal and infantile juvenile form).
PATHOGENESIS |
CLASSIFICATION |
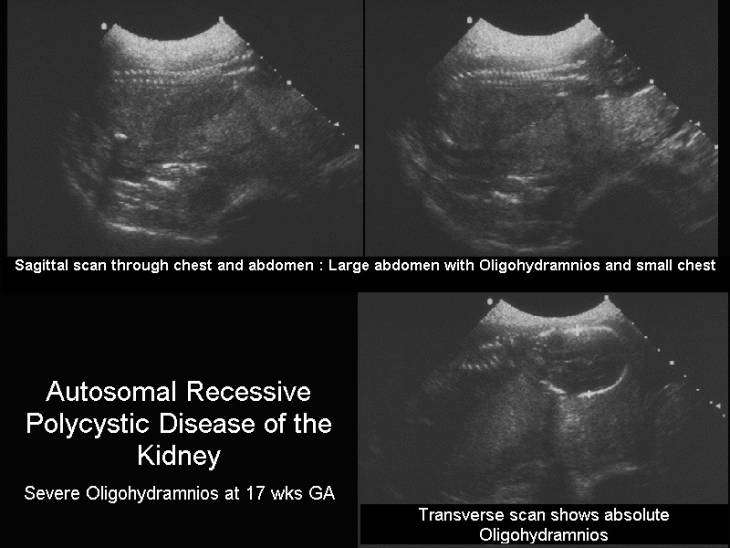
ULTRASOUND |
- Bilateral massively enlarged kidneys (may be seen as early as 16-18 weeks of gestation). A single case of echogenic normal sized kidneys has been reported (2).
- Enlarged kidneys maintain their reniform shape.
- Echogenic (innumerable ectatic collecting tubules result in a multitude of reflective interphases resulting in increased echogenicity).
- Small cysts (1-2mm) may be
distributed throughout the parenchyma, however they are not visualized as
individual structures.
Occasionally 8-10mm cysts may be present. - Severe oligohydramnios (as the kidneys are non functional).
- Absent urinary bladder.
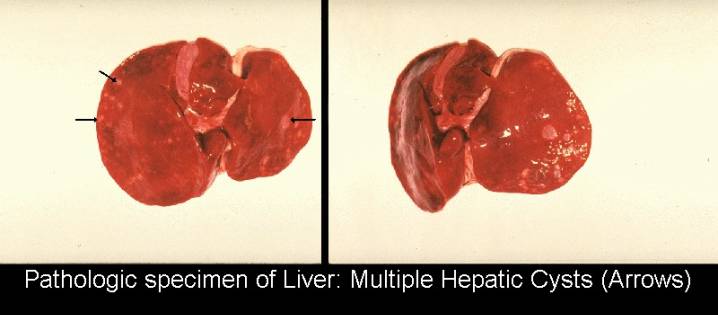
- Hepatic abnormalities (there is an inverse relationship between the degree of hepatic and renal involvement).
- Hepatic cysts.
- Bile duct proliferation.
- Periportal fibrosis (3).
|
|
|
|
|
|
|
|
|
|
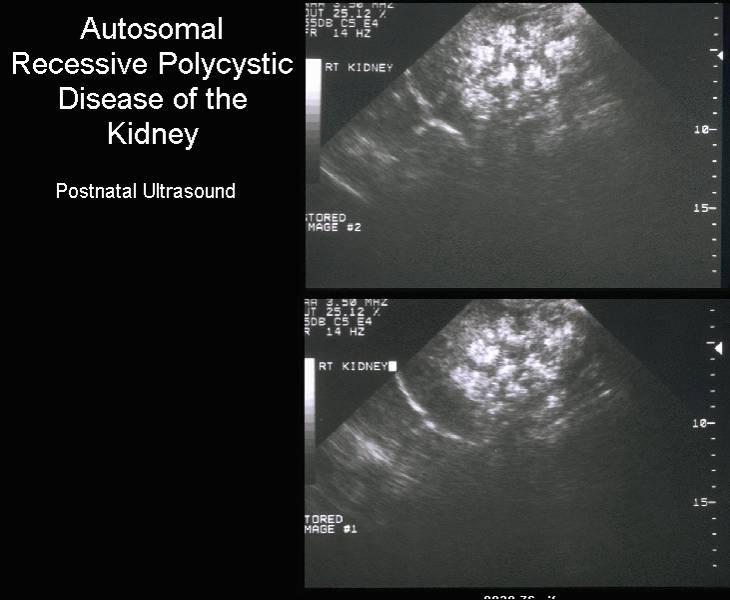
Postnatal
renal ultrasound |
|
|
|
|
|
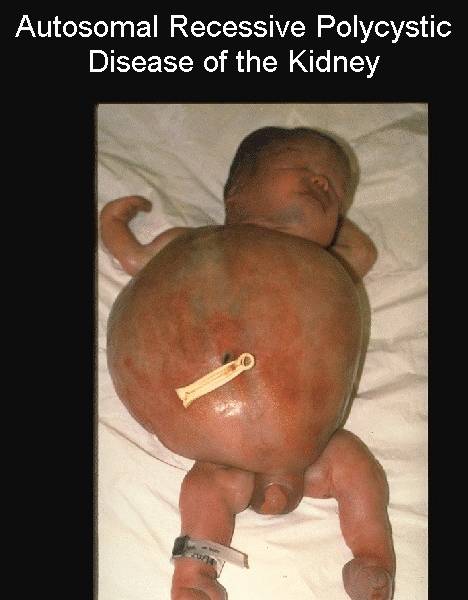
Postmortem
appearance |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Pathological
specimen of kidneys demonstrating the multiple tiny cysts |
|
|
|
|




REFERENCES |
- Chilton SJ, Cremin BJ. The spectrum of polycystic disease in children. Pediatr Radiol 1981;11:9.
- Wisser J, Hebisch G, Froster U et.al. Prenatal sonographic diagnosis of autosomal recessive polycystic kidney disease (ARPKD) during the early second trimester. Prenat Diagn 1995;15:868-871.
- Zerres K, Volpel MC, Weiss H. Cystic kidneys. Genetics, pathologic anatomy, clinical picture and prenatal diagnosis. Hum Genet 1984;68:104-135.